Online first
About the Journal
Current issue
Archive
Publication Ethics
Anti-Plagiarism system
Instructions for Authors
Instructions for Reviewers
Editorial Office
Editorial Board
Contact
Reviewers
All Reviewers
2025
2024
2023
2022
2021
2020
2019
2018
2017
2016
General Data Protection Regulation (RODO)
REVIEW PAPER
New Horizons in Undrestanding and Treating Pompe Disease: Diagnosis and Therapy. A review of the literature.
1
Student’s Scientific Association of Paediatric Neurology, Medical University, Lublin, Poland
2
Department of Children’s Neurology, University Children’s Hospital, Lublin, Poland
Corresponding author
Weronika Zegardło
Student’s Scientific Association of Paediatric Neurology, Medical University of Lublin, Poland
Student’s Scientific Association of Paediatric Neurology, Medical University of Lublin, Poland
J Pre Clin Clin Res. 2024;18(4):303-311
KEYWORDS
TOPICS
ABSTRACT
Introduction and objective:
Pompe disease (PD) is a genetic and metabolic disorder caused by a mutation in the GAA gene, which leads to a deficiency of acid alpha-glucosidase and glycogen deposition in lysosomes. The aim of the study is to review the genetic mechanisms, diagnostic approaches, and potential treatments for PD to improve our understanding and develop effective interventions.
Review methods:
The review examines articles from databases like PubMed, Google Scholar, Clinicaltrials.gov, and NCBI. Meta-analyses, randomized controlled trials, and research articles on PD, diagnosis and therapy were included after initial analysis. More than 95% of the articles are less than eight-years-old.
Brief description of the state of knowledge:
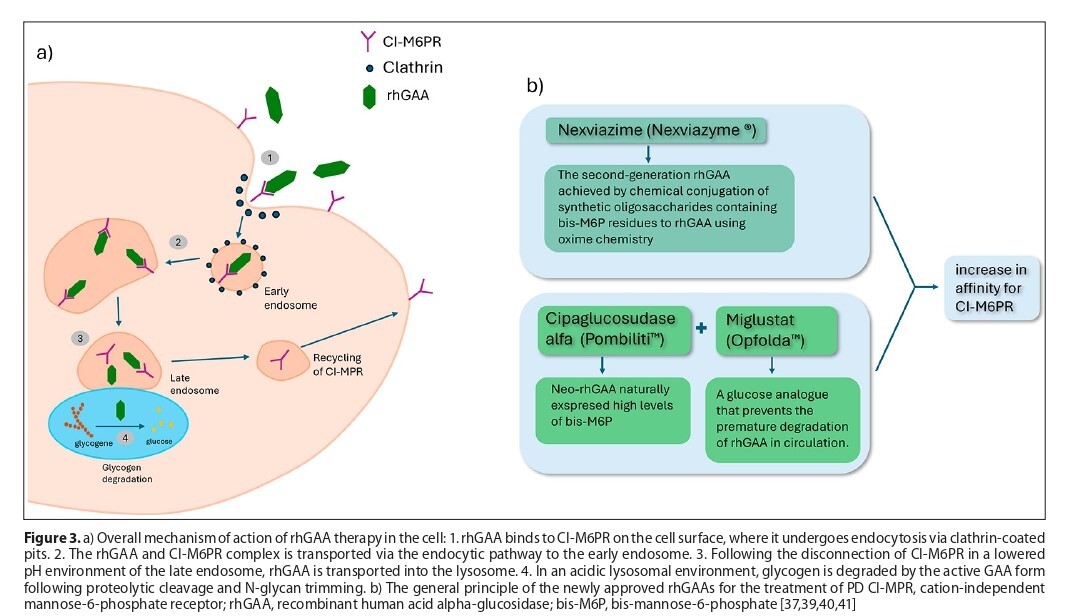
Pompe disease has been a known clinical condition for almost a century, presenting challenges for diagnosis and treatment. Diagnosis can be difficult due to its similarity to other neuromuscular disorders. However, confirmation of the diagnosis can be achieved through enzymology and molecular genetic testing. The current treatment for PD is enzyme replacement therapy using recombinant human alpha-glucosidase. Ongoing research aims to develop improved or new enzymes, as well as other treatments, such as gene therapy and substrate reduction strategies. Early diagnosis and treatment are crucial. Neonatal screening is recommended.
Summary:
Despite advancements in understanding the pathogenesis of PD, the genetic mechanisms underlying its phenotypic variations remain unclear. Reporting cases to databases is crucial for unravelling the molecular basis of symptoms and improving patient outcomes. New therapeutic approaches, such as modified enzyme replacement therapies and gene editing, are essential for overcoming current limitations and improving treatment efficacy in PD.
Pompe disease (PD) is a genetic and metabolic disorder caused by a mutation in the GAA gene, which leads to a deficiency of acid alpha-glucosidase and glycogen deposition in lysosomes. The aim of the study is to review the genetic mechanisms, diagnostic approaches, and potential treatments for PD to improve our understanding and develop effective interventions.
Review methods:
The review examines articles from databases like PubMed, Google Scholar, Clinicaltrials.gov, and NCBI. Meta-analyses, randomized controlled trials, and research articles on PD, diagnosis and therapy were included after initial analysis. More than 95% of the articles are less than eight-years-old.
Brief description of the state of knowledge:
Pompe disease has been a known clinical condition for almost a century, presenting challenges for diagnosis and treatment. Diagnosis can be difficult due to its similarity to other neuromuscular disorders. However, confirmation of the diagnosis can be achieved through enzymology and molecular genetic testing. The current treatment for PD is enzyme replacement therapy using recombinant human alpha-glucosidase. Ongoing research aims to develop improved or new enzymes, as well as other treatments, such as gene therapy and substrate reduction strategies. Early diagnosis and treatment are crucial. Neonatal screening is recommended.
Summary:
Despite advancements in understanding the pathogenesis of PD, the genetic mechanisms underlying its phenotypic variations remain unclear. Reporting cases to databases is crucial for unravelling the molecular basis of symptoms and improving patient outcomes. New therapeutic approaches, such as modified enzyme replacement therapies and gene editing, are essential for overcoming current limitations and improving treatment efficacy in PD.
Zegardło W, Jankowiak K, Kwiatkowski B, Kabała Z, Nowakowska M, Osipowska J, Chrościńska-Krawczyk M, Witas A. New horizons in understanding and treating Pompe disease – Diagnosis and Therapy. A literature review. J Pre-Clin Clin Res. 2024; 18(4): 303–311. doi: 10.26444/jpccr/193653
REFERENCES (50)
1.
Taverna S, Cammarata G, Colomba P, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging (Albany NY). 2020;12(15):15856–15874. doi:10.18632/aging.103794.
2.
Peruzzo P, Pavan E, Dardis A. Molecular genetics of Pompe disease: a comprehensive overview. Ann Transl Med. 2019;7(13):278. doi:10.21037/atm.2019.04.13.
3.
Stevens D, Milani-Nejad S, Mozaffar T. Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Curr Treat Options Neurol. 2022;24(11):573–588. doi:10.1007/s11940-022-00736-1 22.
4.
Hahn A, Schänzer A. Long-term outcome and unmet needs in infantile-onset Pompe disease. Ann Transl Med. 2019;7(13):283. doi:10.21037/atm.2019.04.70.
5.
Labella B, Cotti Piccinelli S, Risi B, et al. A Comprehensive Update on Late-Onset Pompe Disease. Biomolecules. 2023;13(9):1279. Published 2023 Aug 22. doi:10.3390/biom13091279.
6.
Prakash S, Penn JD, Jackson KE, Dean LW. Newborn screening for Pompe disease: Parental experiences and follow-up care for a late-onset diagnosis. J Genet Couns. 2022;31(6):1404–1420. doi:10.1002/jgc4.1615.
7.
El Haddad L, Khan M, Soufny R, et al. Monitoring and Management of Respiratory Function in Pompe Disease: Current Perspectives. Ther Clin Risk Manag. 2023;19:713–729. Published 2023 Sep 1. doi:10.2147/TCRM.S362871.
8.
Herbert M, Case LE, Rairikar M, et al. Early-onset of symptoms and clinical course of Pompe disease associated with the c.-32–13 T > G variant. Mol Genet Metab. 2019;126(2):106–116. doi:10.1016/j.ymgme.2018.08.009.
9.
Nancy Leslie, Laurie Bailey. Pompe Disease. GeneReviews®, edited by Adam MP, et al. University of Washington, Seattle, 31 August 2007.
10.
Pfrimmer C, Smitka M, Muschol N, et al. Long-Term Outcome of Infantile Onset Pompe Disease Patients Treated with Enzyme Replacement Therapy – Data from a German-Austrian Cohort. J Neuromuscul Dis. 2024;11(1):167–177. doi:10.3233/JND-230164.
11.
Kenney-Jung D, Korlimarla A, Spiridigliozzi GA, et al. Severe CNS involvement in a subset of long-term treated children with infantile-onset Pompe disease. Mol Genet Metab. 2024;141(2):108119. doi:10.1016/j.ymgme.2023.108119.
12.
Toscano A, Rodolico C, Musumeci O. Multisystem late onset Pompe disease (LOPD): an update on clinical aspects. Ann Transl Med. 2019;7(13):284. doi:10.21037/atm.2019.07.24.
13.
Jastrzębska A, Kostera-Pruszczyk A. Multisystem presentation of Late Onset Pompe Disease: what every consulting neurologist should know. Neurol Neurochir Pol. 2023;57(2):143–150. doi:10.5603/PJNNS.a2022.0075.
15.
Kroos M, Pomponio RJ, van Vliet L, et al. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum Mutat. 2008;29(6):E13-E26. doi:10.1002/humu.20745.
16.
de Faria DOS, ‘t Groen SLMI, Hoogeveen-Westerveld M, et al. Update of the Pompe variant database for the prediction of clinical phenotypes: Novel disease-associated variants, common sequence variants, and results from newborn screening. Hum Mutat. 2021;42(2):119–134. doi:10.1002/humu.24148.
17.
Momosaki K, Kido J, Yoshida S, et al. Newborn screening for Pompe disease in Japan: report and literature review of mutations in the GAA gene in Japanese and Asian patients. J Hum Genet. 2019;64(8):741–755. doi:10.1038/s10038-019-0603-7.
18.
Kuperus E, van der Meijden JC, In ‘t Groen SLM, et al. The ACE I/D polymorphism does not explain heterogeneity of natural course and response to enzyme replacement therapy in Pompe disease. PLoS One. 2018;13(12):e0208854. Published 2018 Dec 7. doi:10.1371/journal.pone.0208854.
19.
van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017;24(6):768-e31. doi:10.1111/ene.13285.
20.
Chien YH, Hwu WL, Lee NC. Newborn screening: Taiwanese experience. Ann Transl Med. 2019;7(13):281. doi:10.21037/atm.2019.05.47.
21.
Sawada T, Kido J, Nakamura K. Newborn Screening for Pompe Disease. Int J Neonatal Screen. 2020;6(2):31. Published 2020 Apr 5. doi:10.3390/ijns6020031.
22.
Feeney EJ, Austin S, Chien YH, et al. The value of muscle biopsies in Pompe disease: identifying lipofuscin inclusions in juvenile- and adult-onset patients. Acta Neuropathol Commun. 2014;2:2. Published 2014 Jan 2. doi:10.1186/2051-5960-2-2.
23.
Manganelli F, Ruggiero L. Clinical features of Pompe disease. Acta Myol. 2013;32(2):82–84.
24.
Reynolds TM, Tylee K, Booth K, Wierzbicki AS. PATHFINDER Project Collaboration group; Collaborators and research nurses as listed below. Identification of patients with Pompé disease using routine pathology results: PATHFINDER (creatine kinase) study. J Clin Pathol. 2019;72(12):805–809. doi:10.1136/jclinpath-2019-205711.
25.
Willis TA, Hollingsworth KG, Coombs A, et al. Quantitative muscle MRI as an assessment tool for monitoring disease progression in LGMD2I: a multicentre longitudinal study. PLoS One. 2013;8(8):e70993. Published 2013 Aug 14. doi:10.1371/journal.pone.0070993.
26.
Seiler A, Nöth U, Hok P, et al. Multiparametric Quantitative MRI in Neurological Diseases. Front Neurol. 2021;12:640239. Published 2021 Mar 8. doi:10.3389/fneur.2021.640239.
27.
O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne). 2018;9:402. Published 2018 Aug 3. doi:10.3389/fendo.2018.00402. 27.
28.
Tarallo A, Carissimo A, Gatto F, et al. microRNAs as biomarkers in Pompe disease. Genet Med. 2019;21(3):591–600. doi:10.1038/s41436-018-0103-8.
29.
Ditters IAM, Huidekoper HH, Kruijshaar ME, et al. Effect of alglucosidase alfa dosage on survival and walking ability in patients with classic infantile Pompe disease: a multicentre observational cohort study from the European Pompe Consortium. Lancet Child Adolesc Health. 2022;6(1):28–37. doi:10.1016/S2352-4642(21)00308-4.
30.
Bolano-Diaz C, Diaz-Manera J. Therapeutic Options for the Management of Pompe Disease: Current Challenges and Clinical Evidence in Therapeutics and Clinical Risk Management. Ther Clin Risk Manag. 2022;18:1099–1115. Published 2022 Dec 13. doi:10.2147/TCRM.S334232.
31.
Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149(1):89–97. doi:10.1016/j.jpeds.2006.02.035.
32.
Chien YH, Tsai WH, Chang CL, et al. Earlier and higher dosing of alglucosidase alfa improve outcomes in patients with infantile-onset Pompe disease: Evidence from real-world experiences. Mol Genet Metab Rep. 2020;23:100591. Published 2020 Apr 29. doi:10.1016/j.ymgmr.2020.100591.
33.
Khan AA, Case LE, Herbert M, et al. Higher dosing of alglucosidase alfa improves outcomes in children with Pompe disease: a clinical study and review of the literature. Genet Med. 2020;22(5):898–907. doi:10.1038/s41436-019-0738-0.
34.
Ditters IAM, van Kooten HA, van der Beek NAME, van der Ploeg AT, Huidekoper HH, van den Hout JMP. Are Anti-rhGAA Antibodies a Determinant of Treatment Outcome in Adults with Late-Onset Pompe Disease? A Systematic Review. Biomolecules. 2023;13(9):1414. Published 2023 Sep 19. doi:10.3390/biom13091414.
35.
Li C, Desai AK, Gupta P, et al. Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefits of early treatment with enzyme replacement therapy and immune tolerance induction. Genet Med. 2021;23(5):845–855. doi:10.1038/s41436-020-01080-y.
36.
Yang CF, Yang CC, Liao HC, et al. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J Pediatr. 2016;169:174–80.e1. doi:10.1016/j.jpeds.2015.10.078.
37.
Unnisa Z, Yoon JK, Schindler JW, Mason C, van Til NP. Gene Therapy Developments for Pompe Disease. Biomedicines. 2022;10(2):302. Published 2022 Jan 28. doi:10.3390/biomedicines10020302.
38.
U.S National Library of Medicine, database of privately and publicly funded clinical studies; https://clinicaltrials.gov/ (access: 2024.01.29).
39.
Diaz-Manera J, Kishnani PS, Kushlaf H, et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial [published correction appears in Lancet Neurol. 2022 Apr;21(4):e4]. Lancet Neurol. 2021;20(12):1012–1026. doi:10.1016/S1474-4422(21)00241-6.
40.
Blair HA. Cipaglucosidase Alfa: First Approval. Drugs. 2023;83(8):739–745. doi:10.1007/s40265-023-01886-5.
41.
Schoser B, Roberts M, Byrne BJ, et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): an international, randomised, double-blind, parallel-group, phase 3 trial [published correction appears in Lancet Neurol. 2023 Oct;22(10):e11]. Lancet Neurol. 2021;20(12):1027–1037. doi:10.1016/S1474-4422(21)00331-8.
42.
Mingozzi F, High KA. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race Between Clearance, Tolerance, Neutralization, and Escape. Annu Rev Virol. 2017;4(1):511–534. doi:10.1146/annurev-virology-101416-041936.
43.
Smith BK, Martin AD, Lawson LA, et al. Inspiratory muscle conditioning exercise and diaphragm gene therapy in Pompe disease: Clinical evidence of respiratory plasticity. Exp Neurol. 2017;287(Pt 2):216–224. doi:10.1016/j.expneurol.2016.07.013.
44.
Eggers M, Vannoy CH, Huang J, et al. Muscle-directed gene therapy corrects Pompe disease and uncovers species-specific GAA immunogenicity. EMBO Mol Med. 2022;14(1):e13968. doi:10.15252/emmm.202113968.
45.
Hordeaux J, Dubreil L, Robveille C, et al. Long-term neurologic and cardiac correction by intrathecal gene therapy in Pompe disease. Acta Neuropathol Commun. 2017;5(1):66. Published 2017 Sep 6. doi:10.1186/s40478-017-0464-2.
46.
Keeler AM, Zieger M, Todeasa SH, et al. Systemic Delivery of AAVB1-GAA Clears Glycogen and Prolongs Survival in a Mouse Model of Pompe Disease. Hum Gene Ther. 2019;30(1):57–68. doi:10.1089/hum.2018.016.
47.
Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol Ther. 2018;26(3):664–668. doi:10.1016/j.ymthe.2018.01.018.
48.
Stok M, de Boer H, Huston MW, et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Corrects Murine Pompe Disease. Mol Ther Methods Clin Dev. 2020;17:1014–1025. Published 2020 May 4. doi:10.1016/j.omtm.2020.04.023.
49.
van der Wal E, Bergsma AJ, Pijnenburg JM, van der Ploeg AT, Pijnappel WWMP. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32–13T>G GAA Splicing Variant in Pompe Disease. Mol Ther Nucleic Acids. 2017;7:90–100. doi:10.1016/j.omtn.2017.03.001.
50.
Muñoz S, Bertolin J, Jimenez V, et al. Treatment of infantile-onset Pompe disease in a rat model with muscle-directed AAV gene therapy. Mol Metab. 2024;81:101899. doi:10.1016/j.molmet.2024.101899.
| eISSN: | 1898-7516 |
| ISSN: | 1898-2395 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.